Haematology Watch, Vol 2, Issue 2.
CASE RECORDS
A young girl with menorrhagia and prolonged bleeding time
Mehmood-ul-Hasan
Presentation
A young girl of 20 presented with menorrhagia for the last 2 weeks. She also complained of small red spots on body occuring off and on since her childhood. She was otherwise healthy looking. One of her brothers had history of off and on epistaxis. She also told about off & on epistaxis since childhood. She had a reaction to Aspirin once.
Her symptoms are showing mucosal plus cutaneous bleed, most likely a feature of a disease affecting platelets.
Her general physical examination revealed a comfortably sit well-looking young girl who was well-oriented in time, place & person having Pulse: 84/min, B.P.: 110/70 mm Hg, and Temp.: 98 oF. There was no Pallor and Jaundice. She had no any bruises or petechiae. Systemic examination was unremarkable.
Her blood loss appears to be either small or compensated. And there is no evidence of current cutaneous bleed.
Tabe 1.1 Possible causes of Mucosal bleed
1. PLATELET DISORDERS:
Quantitative disorders (Thrombocytopenia):
Immune thromocytopenia pupura
Liver disease
Heparin
Thrombotic thrombocytopenic purpura
Splenomegaly
Massive transfusion
Disseminated intravascular coagulation
Acute leukaemia
Qualitative disorders:
Uraemia
Myeloproliferative neoplasms
Myelodysplastic disorders
Paroxysmal nocturnal haemoglobinuria
Glanzmann's thrombasthenia
Bernard-Soulier syndrome
Storage pool diseases
2. COAGULATION DISORDERS:
Vitamin K deficiency
Anticoagulants
3. LOCAL LESIONS:
Leiomyoma uteri
Vasculitis
She was admitted and the physician started Tab. DUPHASTON (Dydrogesterone) 10 mg TDS, Cap. TRANSAMIN (Tranexamic acid) 500 mg TDS, and Tab. FOLIC ACID 5 mg TDS. Her routine tests were sent to the laboratory.
Her CBC showed Hb: 9 g/dL [11.5 - 15 g/dL], Platelets: 14 000/μL [150 000 - 400 000/μL], WBC: 4 000/uL [4 000 – 11 000/μL], MCV: 75 fL [83 – 101 fL], MCH: 23 pg [27 – 31 pg]. The Peripheral Blood Morphology showed Microcytic hypochromic anaemia and Thrombocytopenia corresponding to ~ 70 000/μL with giant forms; no platelet clumps were seen. No other abnormal cells were seen.
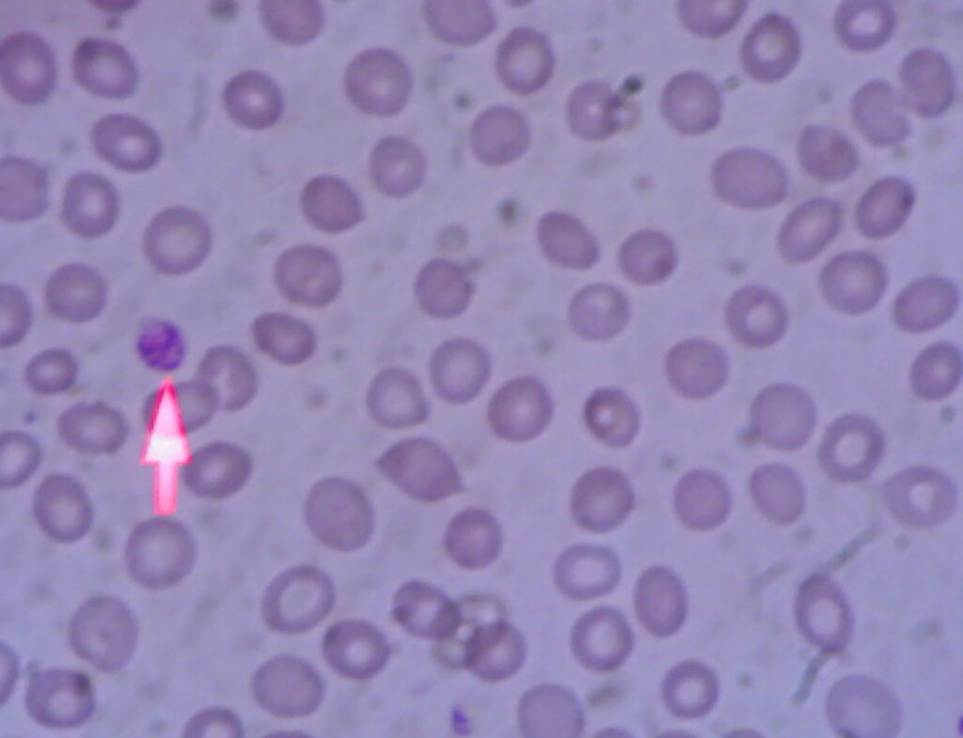
Bicytopenia is seen. A corrected platelet count of 70 000/μL appears not be the reason of bleed until there is a coexistent functional defect.
Further results showed Prothrombin time: 15/14 sec, APTT: 30/29 sec, Bleeding time: Not done due to platelet # < 50 000/μL, ALT: 20 U/L [< 37 U/L], Alkaline Phosphatase: 208 [65 - 306 IU/L ], Creatinine: 90 μmol/L [60 – 156 μmol/L]. After corrected platelet count, her Bleeding time was requested,that came out to be 12 min [2-7 min]. USG Abdomen showed Adnexal mass Rt. Side, a suspected Ovarian cyst.
Her baseline coagulation tests were normal except a prolonged bleeding time. Her platelet function studies can help.
Her platelet function tests were advised. These showed Platelet count: 40 × 109/L [150 – 400 × 109/L], Bleeding time: 12 min [2 – 7 min], Platelet aggregation tests showed 1. Collagen= Normal response, 2. ADP= Normal response, 3. Epinephrine= Normal response, 4. Ristocetin= No response, 5. Ristocetin (Pt PRP+control PPP)=No response. Hess’ test was negative.
A diagnosis of Bernard-Soulier syndrome was made. She was discharged on Tab. TRANSAMIN (Tranexamic acid) 500 mg, TDS, as long as bleeding continues; Tab. PRIMOLUT-N (Norethisterone) 5mg, 1 TDS, Tab. FOLIC ACID 1 BID, and DDAVP (1-desamino-8-D-arginine vasopressin) 0.3 mg/kg S/C.
DISCUSSION
Bernard-Soulier Syndome
It is an inherited disorder characterized by thrombocytopenia, giant platelets, and a failure of the platelets to undergo selective von Willebrand factor-dependent platelet interactions due to abnormalities in the platelet GPIb/IX complex.
In 1948, BERNARD AND SOULIER described a young male patient with a severe bleeding disorder that was characterized by a prolonged bleeding time, thrombocytopenia, and extremely large platelets. It is always transmitted in an autosomal recessive manner and often occurring in persons whose parents are close relatives.
In 1969, Gröttum and Solum, noted reduced electrophoretic mobility of the platelets due to a marked decrease in the concentration of sialic acid on their membranes.Howard et al and Caen and Levy-Toledano found that the platelets of BSS patients failed to aggregate to ristocetin, a peptide antibiotic known to aggregate normal platelets but not the platelets of patients suffering from vWD.
In 1974, Weiss et al demonstrated a defect in the ability of BSS platelets to adhere to rabbit aortic subendothelium. They also suggested that the defect resulted from absence of a receptor for vWF on the platelet surface.
The importance of this receptor for normal hemostasis is perhaps best illustrated by the clinical history of the original patient described by Bernard and Soulier. As both a child and a young man, this patient suffered numerous bleeding problems, including prolonged bleeding after tooth extraction, life-threatening cerebrospinal hemorrhage, and orbital and periorbital hematomas after an accident. At 28 years of age, he died of intracranial hemorrhage after a barroom brawl.
The biochemical defect was defined further in the laboratories of Clemetson et al and Berndt et al, when they demonstrated, in unrelated patients with BSS, deficiencies of 4 polypeptides: glycoproteins (GP) Ib, Ib, IX, and V. These polypeptides all associate on the platelet surface to form a receptor called the GP Ib-IX-V complex.
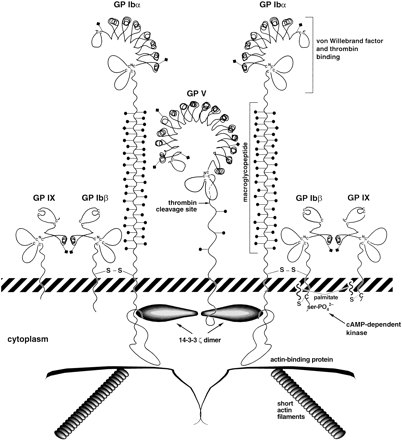
The principal function of the GP Ib-IX-V complex in hemostasis is to initiate the arrest of platelets at sites of vascular injury. Like other adhesion receptors, ligation of the GP Ib-IX-V complex by vWF can transduce signals to the platelet cytoplasm, initiating the cascade of events that leads to the formation of a hemostatic platelet plug.
The platelets are larger than normal and they do not aggregate with ristocetin.