{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Paroxysmal nocturnal hemoglobinuria (PNH), is an acquired clonal abnormality of pluripotent haemopoietic stem cell due to dominant somatic loss-of-function mutations in PIG-A gene at Xp22.1 leading to absence of N-acetyl GPI anchor resulting in low CD59±CD55, CD66b, high complement-mediated haemolysis, and a propensity for venous thrombosis and bone marrow suppression.
Sometimes referred to as Marchiafava-Micheli syndrome, it is a rare and potentially life-threatening disease of the blood characterised by complement-induced intravascular haemolytic anaemia (anemia due to destruction of RBCs in the bloodstream), red urine (due to the appearance of haemoglobin in the urine) and thrombosis.
PNH is the only hemolytic anemia caused by an acquired (rather than inherited) intrinsic defect in the cell membrane (deficiency of glycophosphatidylinositol leading to absence of protective proteins on the membrane).
History
The first description of paroxysmal hemoglobinuria was by the German physician Paul Strübing 1882. A more detailed description was made by Dr Ettore Marchiafava and Dr Alessio Nazari in 1911, with further elaborations by Marchiafava in 1928 and Dr Ferdinando Micheli in 1931. The Dutch physician Enneking coined the term "paroxysmal nocturnal hemoglobinuria" (or haemoglobinuria paroxysmalis nocturia in Latin) in 1928.
Clinical Features
Many, but not all, patients have cola-coloured urine at some point in their disease course. Many of them continue to have low-grade breakdown of red blood cells, leading to typical symptoms of anemia. On laboratory examination of the urine, breakdown products of red blood cells (haemoglobin and haemosiderin) may be identified. A small proportion of patients report abdominal pain, dysphagia (difficulty swallowing), as well as erectile dysfunction in men - this occurs mainly when the breakdown of red blood cells is rapid.
Many patients develop thrombosis at some point in their illness. This is the main cause
of severe complications and death in PNH. These may develop in common
sites e.g. leg veins and resultant pulmonary embolism , but, in PNH, blood clots may also form in more unusual sites e.g. the hepatic vein (causing Budd-Chiari Syndrome), the portal vein of the liver, the mesenteric veins, and veins of the skin.
Diagnosis
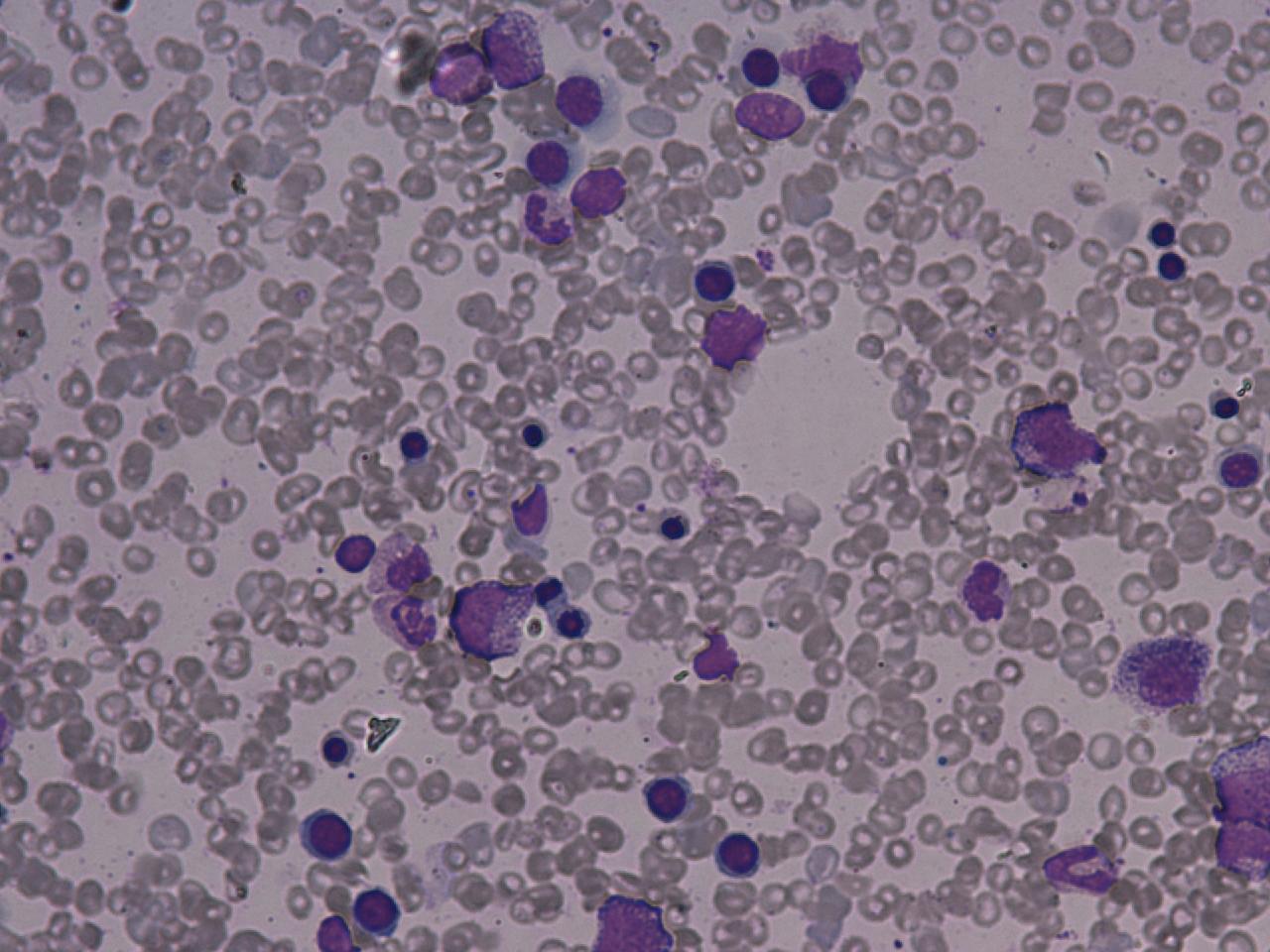
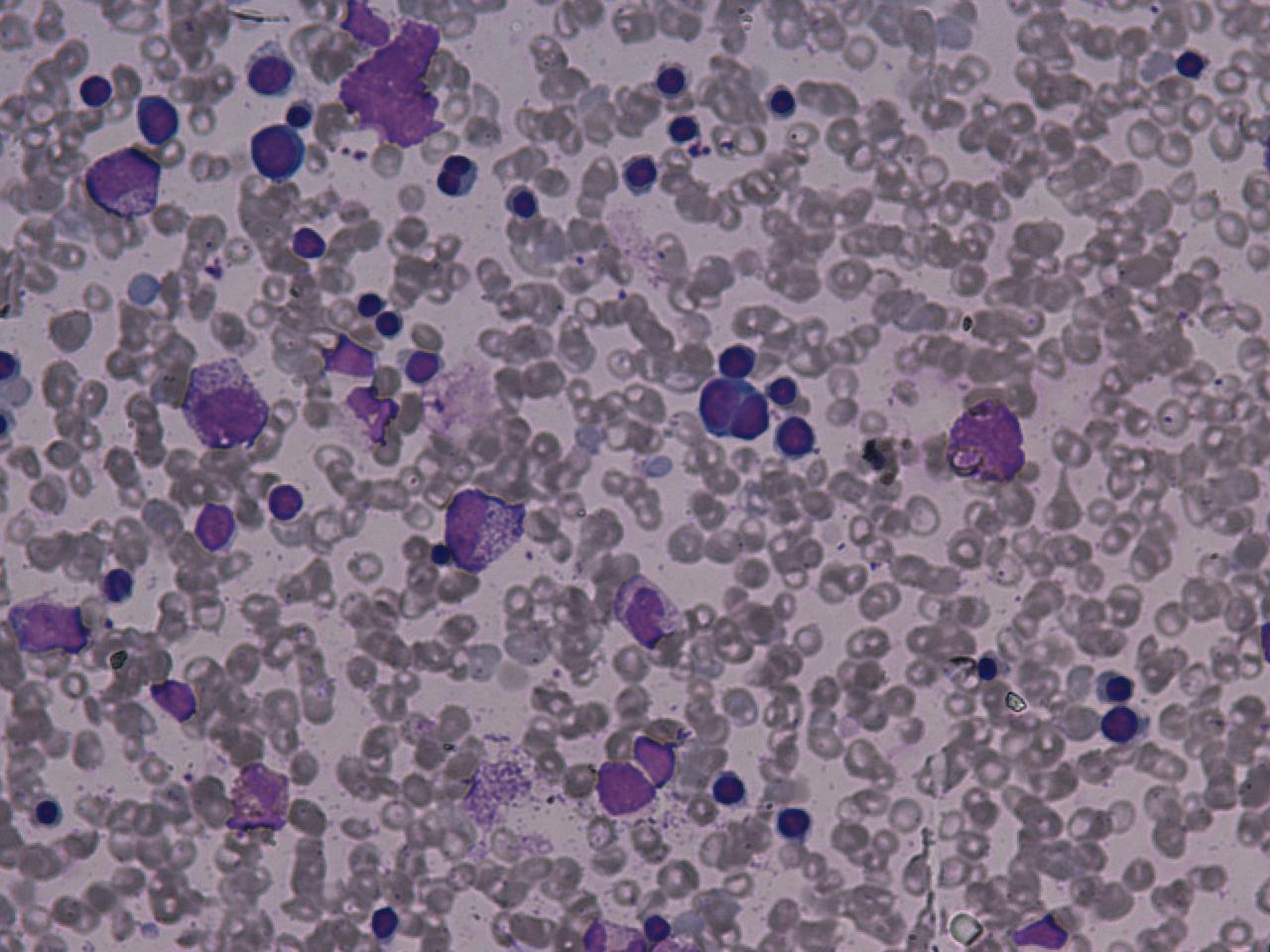
Results of blood tests in PNH are consistent with intravascular haemolytic anaemia: low haemoglobin, raised LDH, high reticulocytes, raised unconjugated bilirubin (a breakdown product of hemoglobin), and decreased levels of haptoglobin. The DAT (direct Coombs' test) is negative, as the hemolysis of PNH is not caused by antibodies.
Historically, the sucrose lysis test, in which a patient's red blood cells are placed in low-ionic-strength solution and observed for hemolysis, was used for screening. If this was positive, the Ham's acid hemolysis (after Dr Thomas Ham, who described the test in 1937) test was performed for confirmation.
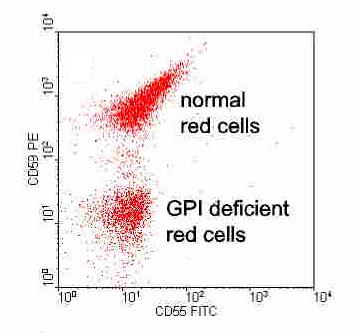
Today, many labs use flowcytometry for CD55 (Complement decay accelerating factor) and CD 59 (Membrane inhibitor of reactive lysis) on WBCs and RBCs.
Based on the levels of these cell proteins, erythrocytes may be
classified as type I, II, or III PNH cells. Type I cells have normal
levels of CD55 and CD59; type II have reduced levels; and type III have
absent levels. It should be noted that as these patients are frequently transfused by RCC, detection of CD 55 & CD59 on WBCs is more accurate method, and correlates well with marrow clone size.
The Fluorescein-labeled proaerolysin (FLAER) test is being used more frequently to diagnose PNH. FLAER binds selectively to the glycophosphatidylinositol anchor and is more accurate in demonstrating a deficit than flow cytometry for CD59 or CD55.
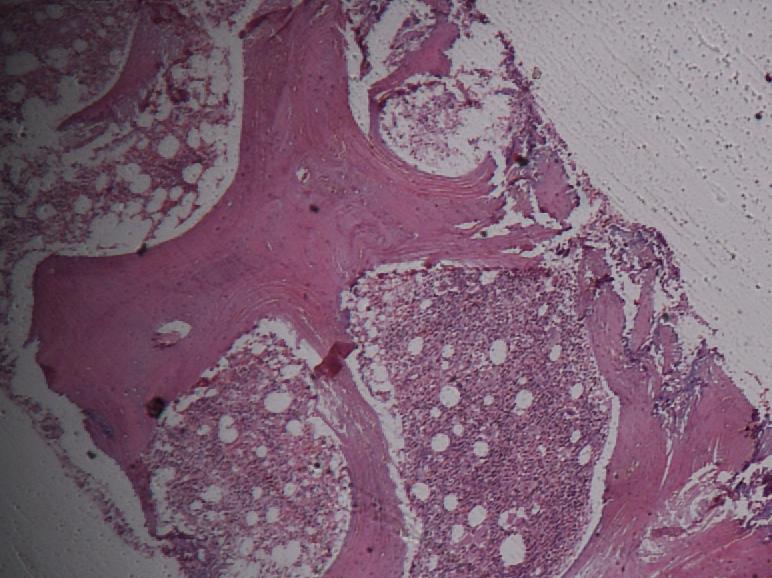
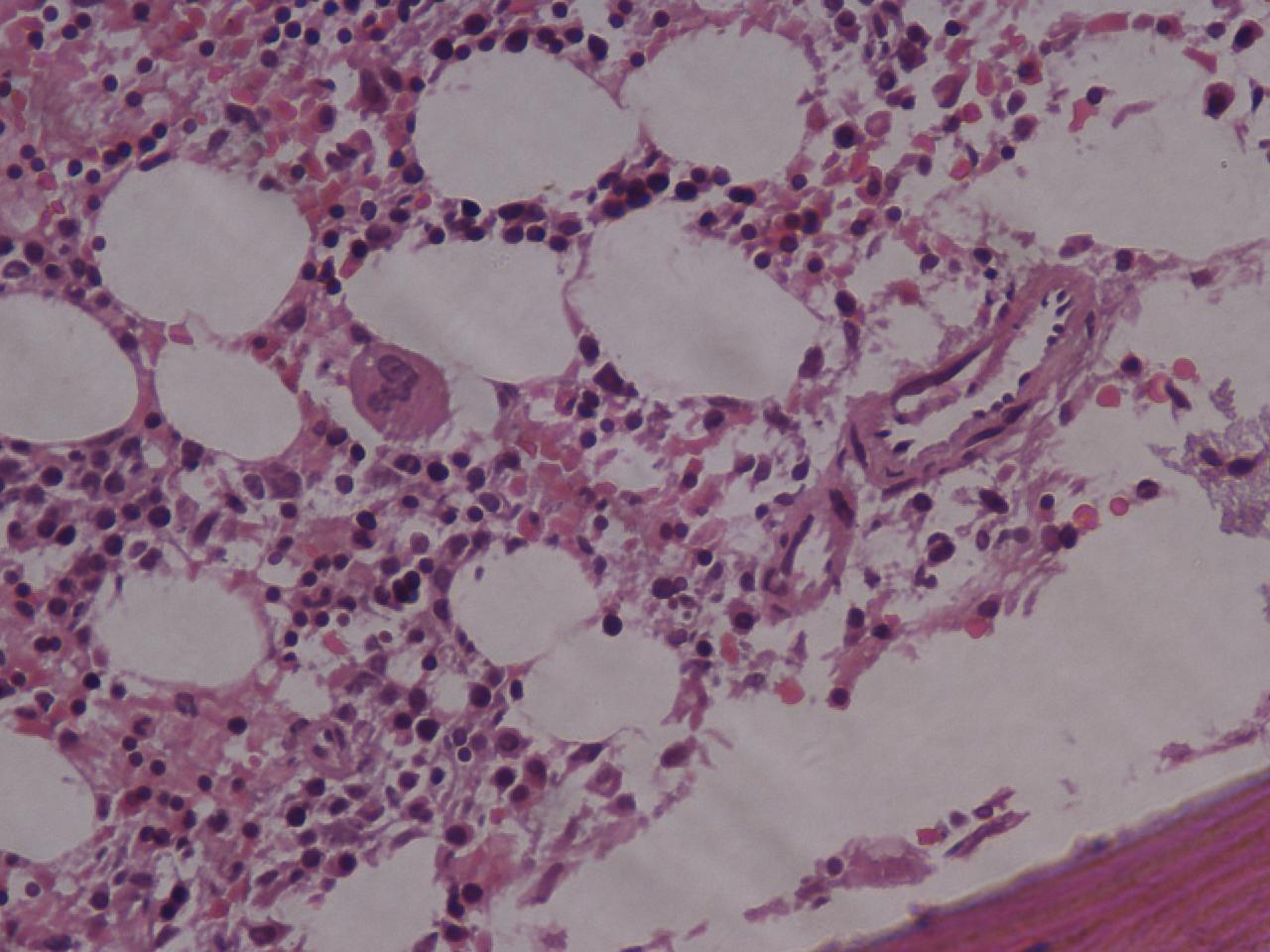
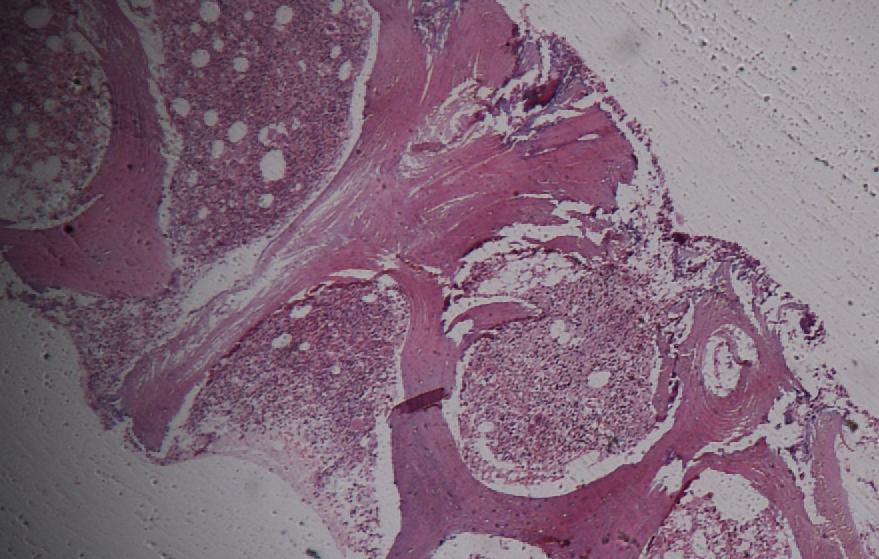
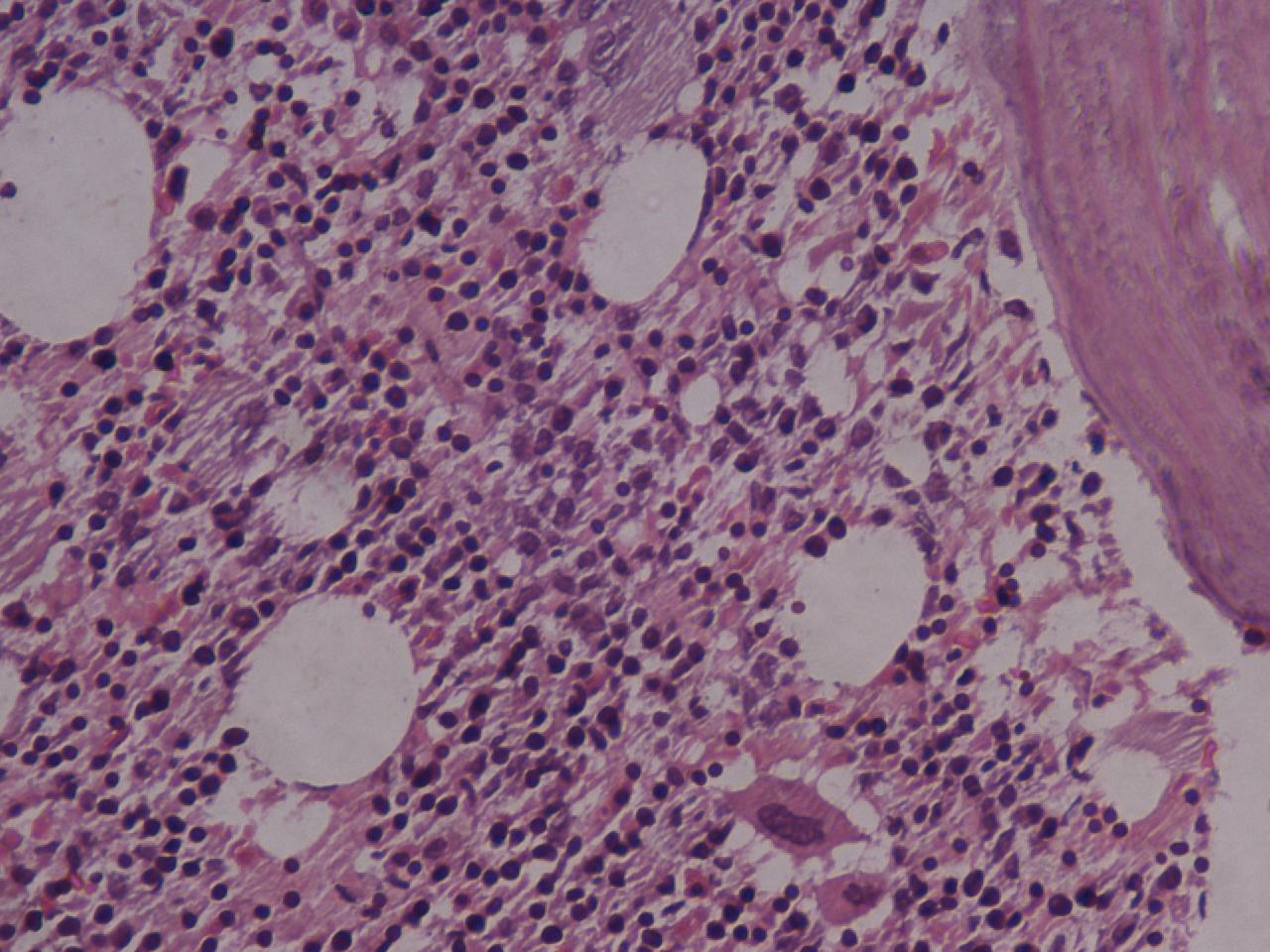
Pathology
All cells have proteins attached to their membranes that are responsible for performing a vast array of functions. There are several ways for proteins to be attached to a cell membrane. PNH occurs as a result of a defect in one of these mechanisms.
The enzyme phosphatidylinositol-A (PIGA) is needed to make glycophosphatidylinositol (GPI), a molecule that anchors proteins to the cell membrane. The gene that codes for PIGA is located on the X chromosome, which means that only one active copy of the gene for PIGA is present in each cell (initially, females have two copies, but one is silenced through X-inactivation). If a mutation occurs in this gene then PIGA may be defective, which leads to a defect in the GPI anchor. When this mutation occurs in a bone marrow stem cell (which are used to make red blood cells as well as white blood cells and platelets), all of the cells it produces will also have the defect. Several of the proteins that anchor to GPI on the cell membrane are used to protect the cell from destruction by the complement system, and, without these anchors, the cells are more easily targeted by the complement proteins. The complement system is part of the immune system and helps to destroy invading microorganisms. Without the proteins that protect them from complement, red blood cells are destroyed. The main proteins that carry out this function are the Decay-accelerating factor (DAF) (CD55), which disrupts formation of C3 convertase, and Protectin/Membrane inhibitor of reactive lysis (CD59), which binds the Membrane attack complex and prevents CD9 from binding to the cell.
The increased destruction of red blood cells results in anemia. The increased rate of thrombosis is due to dysfunction of platelets due to binding by complement, or possibly due to low nitric oxide levels.
Management
There is disagreement as to whether steroids (such as Prednisolone) can decrease the severity of haemolysis. Transfusion therapy may be needed; in addition to correcting significant anaemia, this suppresses the production of PNH cells by the bone marrow, and indirectly the severity of the hemolysis. Iron deficiency develops with time, due to losses in urine, and may have to be treated if present. Iron therapy can result in more hemolysis as more PNH cells are produced.
PNH is a chronic condition. In patients with only a small clone and few problems, monitoring of the flow cytometry every six months gives information on the severity and risk of potential complications. Given the high risk of thrombosis in PNH, preventative treatment with Warfarin decreases the risk of thrombosis in those with a large clone (50% of white blood cells type III).
Episodes of thrombosis are treated as they would in other patients, but, given that PNH is a persisting underlying cause, it is likely that treatment with Warfarin or similar drugs needs to be continued long-term after an episode of thrombosis. Allogeneic bone marrow transplantation is the only curative therapy, although the monoclonal anti-complement antibody Eculizumab (Soliris) is effective at reducing the need for blood transfusions, improving quality of life, and reducing the risk of thrombosis.
Our patient had no thrombotic symptoms and a nutrtional deficiency was evident due to continuous low-grade intravascular haemolysis.